欧盟GMP检查:从查询途径到检查趋势分析
2023-10-10 10:14:00
今年以来,GMP现场检查如火如荼地展开,特别是FDA对国外制药企业的检查次数明显增加,引发行业内都在关注FDA 483和警告信,相应的“483报告”和警告信也颇具看点。但是,今天我们将不再深入讨论FDA的情况,而是来关注欧盟近些年来对欧洲经济区及第三国的GMP检查情况。那么关于欧盟的GMP检查结果我们可以在哪里查询?近些年来欧盟的检查趋势如何发展?什么情况下会发布GMP不合格报告?
在这篇文章中,我们将一一探讨这些问题,帮助您更好地了解欧盟的GMP检查现状。
EudraGMDP介绍
在欧盟人用药品2004/27/EC 号指令和兽用药品 2004/28/EC 号指令中引入了“共同体(Community)数据库”的法律框架,其中规定了成员国在GMP检查后需要将其颁发的GMP证书和GMP不符合性声明输入到数据库中,这里的“数据库”也就是EudraGMDP(http://eudragmdp.ema.europa.eu/inspections/displayWelcome.do)。EudraGMDP从2011年开始可供公众查询,在该数据库中我们可以查询到EEA及第三国药品及生产商的以下内容:
-
生产和进口许可(Manufacturing and import authorisations)
-
GMP证书(Good Manufacturing Practice (GMP) certificates.)
-
GMP不符合性声明(Statements of non-compliance with GMP)
-
第三国的GMP检查计划(GMP inspection planning in third countries)
-
批发分销许可(Wholesale Distribution Authorisations)
-
GDP证书(Good Distribution Certificates (GDP))
-
GDP不符合性声明(Statements of non-compliance with GDP)
-
EEA人用活性物质制造商、进口商和分销商注册(Registration of manufacturers, importers and distributors of active substances for human use located in the EEA)
下面我们将重点介绍一下GMP certificates和Statements of non-compliance with GMP。
在GMP检查后,执行检查的机构负责在90天内将证书的详细信息输入EudraGMDP。这些负责检查的机构包括了EC、EMA、EDQM、成员国的药监机构,如果是联合检查,那么这些机构也会确定由谁来签发证书。
没有经历过欧盟检查的小伙伴经常会问我们,欧盟的GMP证书长啥样,GMP不符合性声明会发什么内容?GMP证书和GMP不符合性声明的模板可以在EMA发布的文件中找到:Compilation of Union Procedures on Inspections and Exchange of Information,Rev 19.1(PS.这份文件是欧盟检查和信息交流的汇编,包括了GMP/GDP检查报告、注册和GMP/GDP证书等的模板以及说明,最新版的在2023.09.15更新,全文有297页,信息丰富,先挖个坑,后续可以选择部分章节跟大家分享交流)。此处截取了GMP证书首页(图一)和尾页(图二)的信息如下图所示:


2.2 GMP不符合性声明
欧盟GMP不符合声明与FDA的警告信相同的是,他们都有一定的强制性,具有制裁力;不同的是:FDA警告信是针对检查现场发现的违法行为的警示和劝告,内容更详细,公布与众的一方面是警告被检查企业,另一方面是警示其他企业有则改之无则加勉。但欧盟的GMP不符合性声明更简单明了,告诉你有几条不符合项,来自于哪些地方,官方会对你采取哪些行动,例如:收回你的GMP证书,禁止你的产品销往欧盟。
GMP不符合性声明分为三个部分:
Part 1:公司的基本信息及违反的法规;
在这一部分,会根据本次检查的范围,填写违反的法规,不同药品适用的法规如下:
人用药:Directive (EU) 2017/1572
IMP:Regulation (EU) 2017/1569
兽药:Directive 91/412/EEC
API:Directive 2001/83/EC
兽药API:Regulation (EU) 2019/6

Part 2:与本NCS相关的制造商的活动;
根据本次检查的范围,选择模块中对应内容,针对人用药品、兽用药品、临床试验用药三个部分,包括的模块有:无菌产品、非无菌产品、生物制品、包装、质量控制等,对于进口药品也有进口质量控制检测和批放行(Batch certification of imported medicinal products)等对应选项,如没有列举的内容,需要在“Other”中补充。

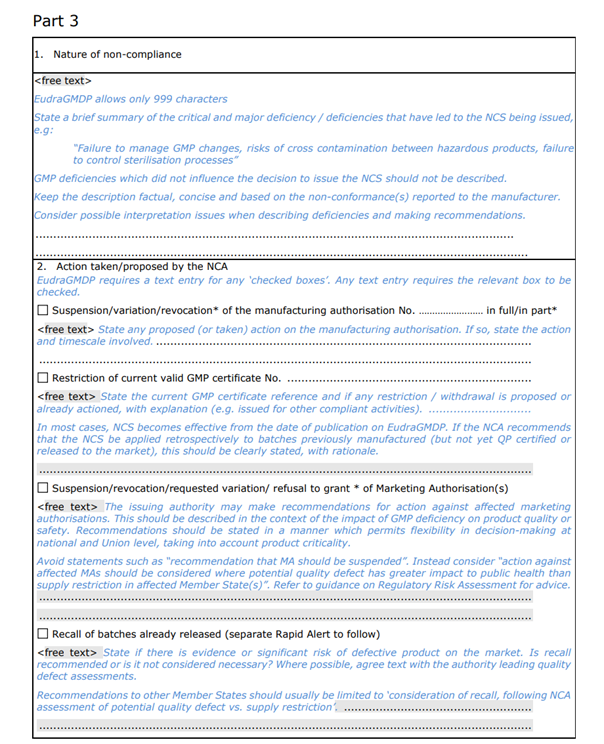
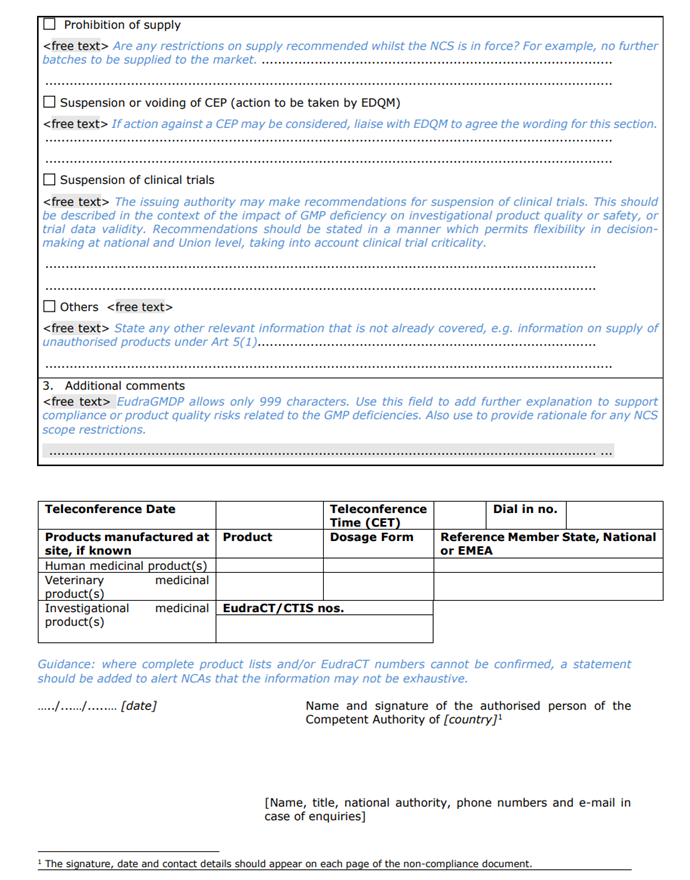
Part 3:不符合项目的说明及官方所要采取的措施;
声明中仅概括了导致本次NCS的关键(critical)和主要(major)缺陷,包括缺陷的数量及简单描述,并未详细的描述具体如何违反GMP。NCA(National Competent Authorities)已采取或建议采取的行动,包括撤销/变更/暂停生产许可、GMP证书、上市许可,中止CEP证书,中止临床试验等情况。
通过欧盟GMP检查企业的缺陷项并未包括在EudraGMDP中,官方会不定期的发布和总结,例如EDQM在2019发布的2006~2018检查和缺陷趋势概况文件(EDQM inspections and trends of deficiencies Overview 2006 to 2018),这份文件中就包括2006~2018年API检查及缺陷分析,大家可以在网上自行查询。
2017~2023年EU GMP检查数据分析
我们整理了2017.01.01至今(2023.09.15)EU GMP检查的四组数据,即在全世界的检查总数、第三国检查数量、在中国的检查数量以及发布的GMP不符合声明数量(注:这里的检查数量包括了发放GMP证书和未通过GMP检查的总量,如还有其他可能性,不包括在内)。
制作了两份图表来对比,图一为不同年份及对应的检查数量,图二是不同年份第三国检查数量、中国检查数量以及发布的不符合声明数量的占比。
如果我们假设检查官都会在检查完成后90天才登记在数据库中,那么2023年的数据刚好是6个月,可以代表2023年一半的检查量。
仁者见仁,各位看官对这些数据有何看法?
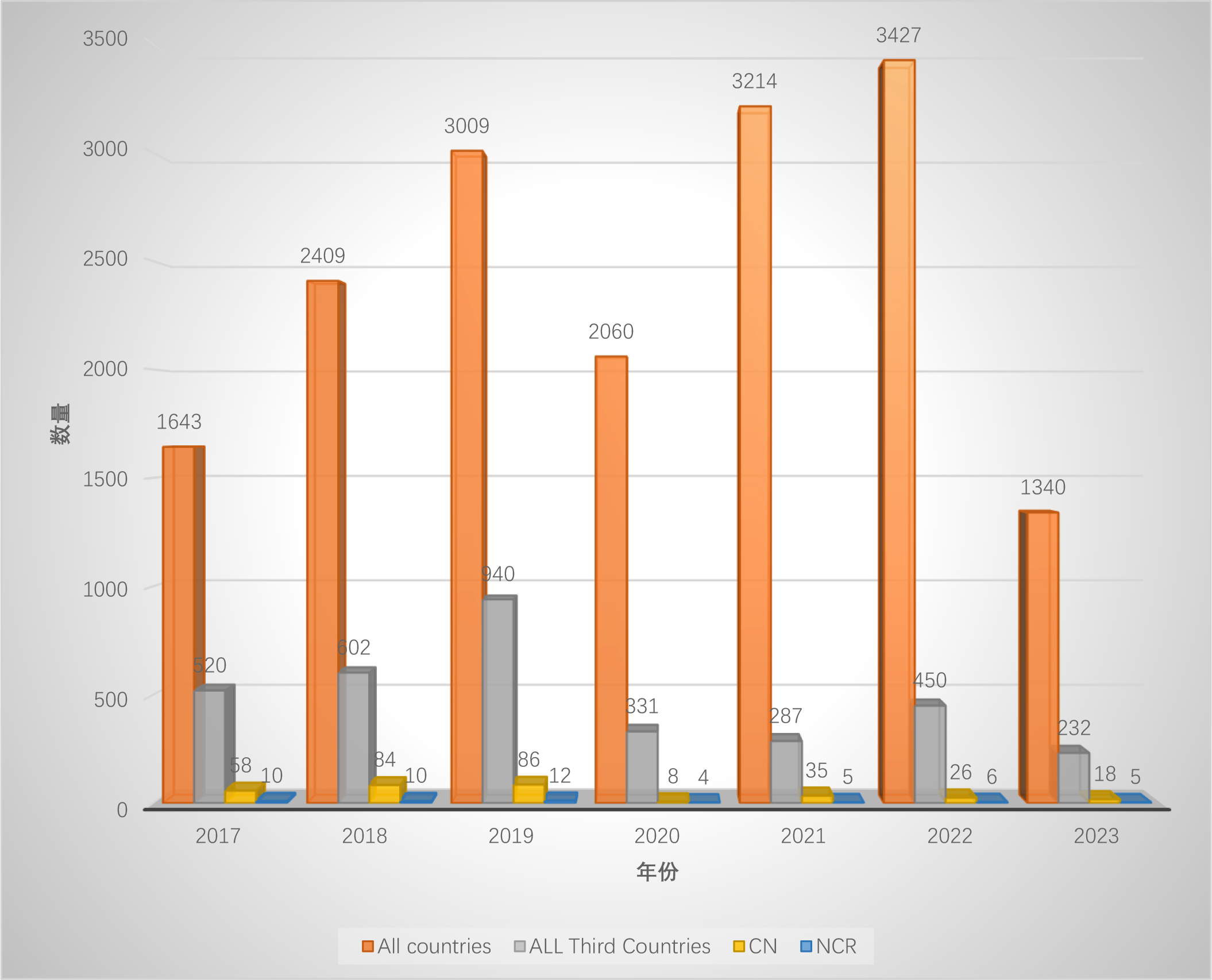
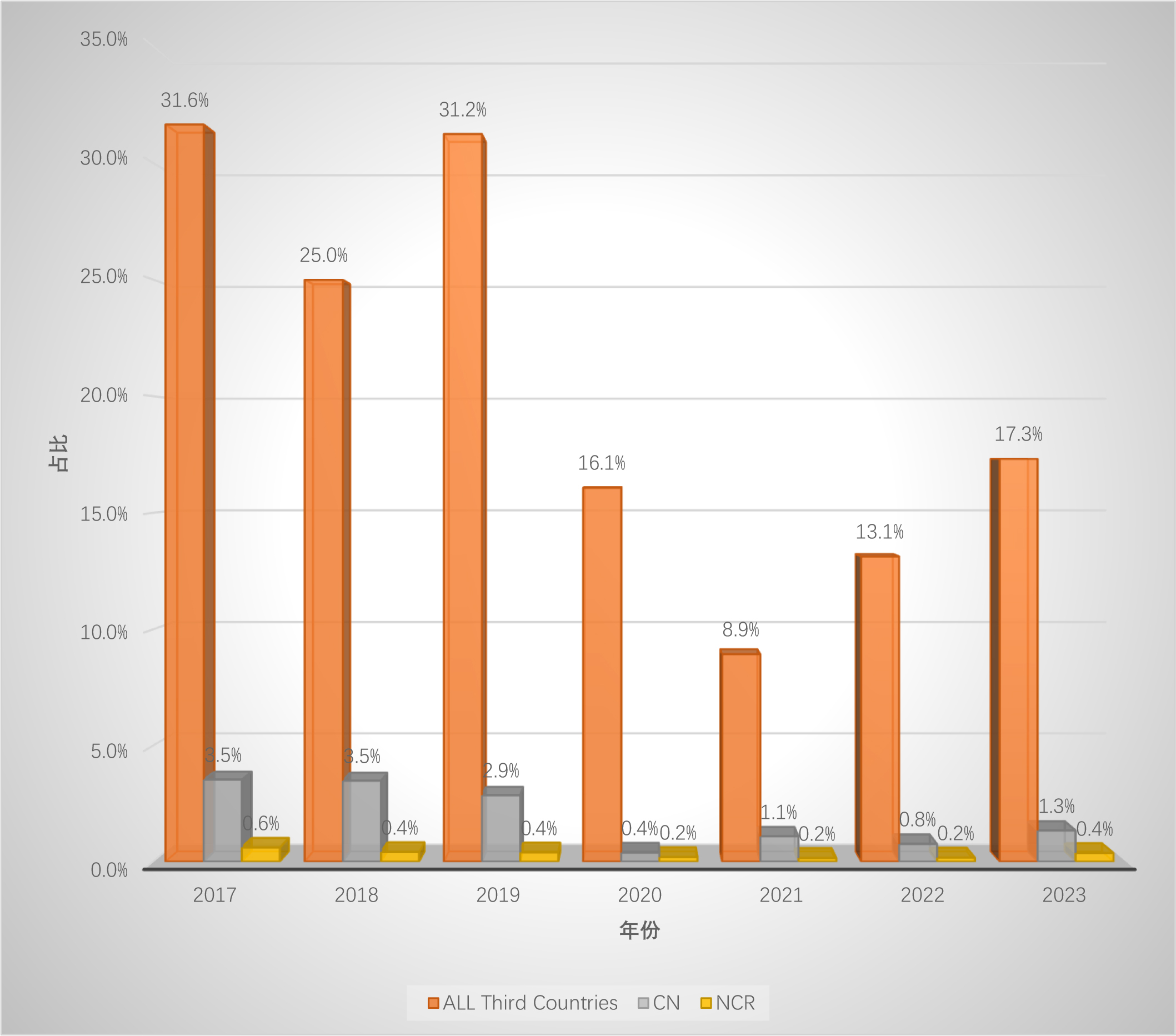
从近些年欧盟 GMP检查数据来看,国内药企接受欧盟GMP检查的数量并没有我们想象中的多,相较于FDA,欧盟的注册费用并不高,对想要走上国际化增加竞争力的药企,欧盟或许是个不错选择。康利华在原辅料、化学药品、生物制品等医药产品领域持续提供专业的欧盟EU GMP咨询服务,助力各类医药公司及制药企业应对EDQM、EMA及成员国的现场或远程检查、QP审计等;
-
EU GMP差距分析
-
质量体系提升
-
专题培训
-
模拟审计
-
迎检准备
-
现场陪检及翻译
-
在下一期的文章中,我们计划讨论欧盟近年来发布的GMP不符合性声明,研究了解这些声明中究竟涉及了哪些缺陷,以及是什么原因导致了检查未通过。我们欢迎各位继续关注,共同学习,共同进步。
策划:魏巍
编写:王亚蕊









