







国际药品注册事务
美国DMF申请
为全球生物科技、医药公司和制药企业提供注册事务服务
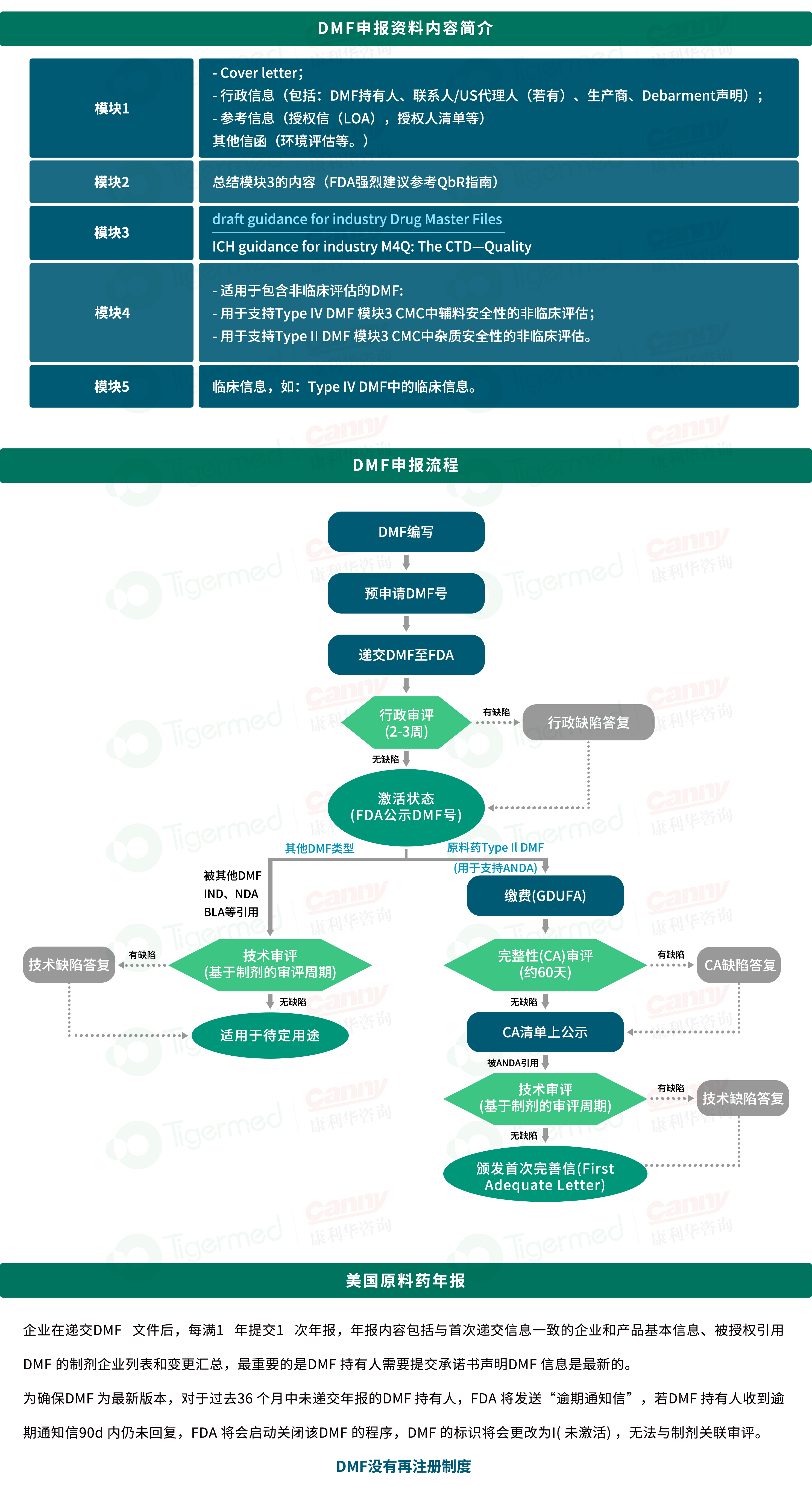
DMF,即 Drug Master File,直译为 “药品主文件” 。它是美国 FDA(食品药品监督管理局)要求的一份用于提交药品、活性成分、辅料、包装材料等相关数据的非公开文件。DMF分类:DMF 共分为五种类型,每种类型对应不同的产品或信息类别:
1. Type I:生产设施、设备和运行场所的信息(目前很少使用)。
2. Type II:药物活性成分(API)、制剂或中间体的制造、加工或包装信息。大多数情况下,企业提交的是 Type II DMF,用于 API 或原料药的注册。
3. Type III:包装材料的信息,像各类药品包装所用的容器等信息都包含在内,其对保障药品稳定性、安全性意义重大。
4. Type IV:辅料、配方或着色剂的信息。
5. Type V:FDA 要求的特定信息(仅在 FDA 批准时使用) 。
精准定位 DMF 类型,既能确保企业提交的资料有的放矢,又能加速后续的审评流程。
全国热线:
400-8770626
邮箱:
Canny@TigermedGrp.com
地址:
中国·北京市朝阳区朝阳门外大街20号联合大厦808室
邮编:
100022

