







国内药品注册事务
eCTD格式转换
为全球生物科技、医药公司和制药企业提供注册事务服务
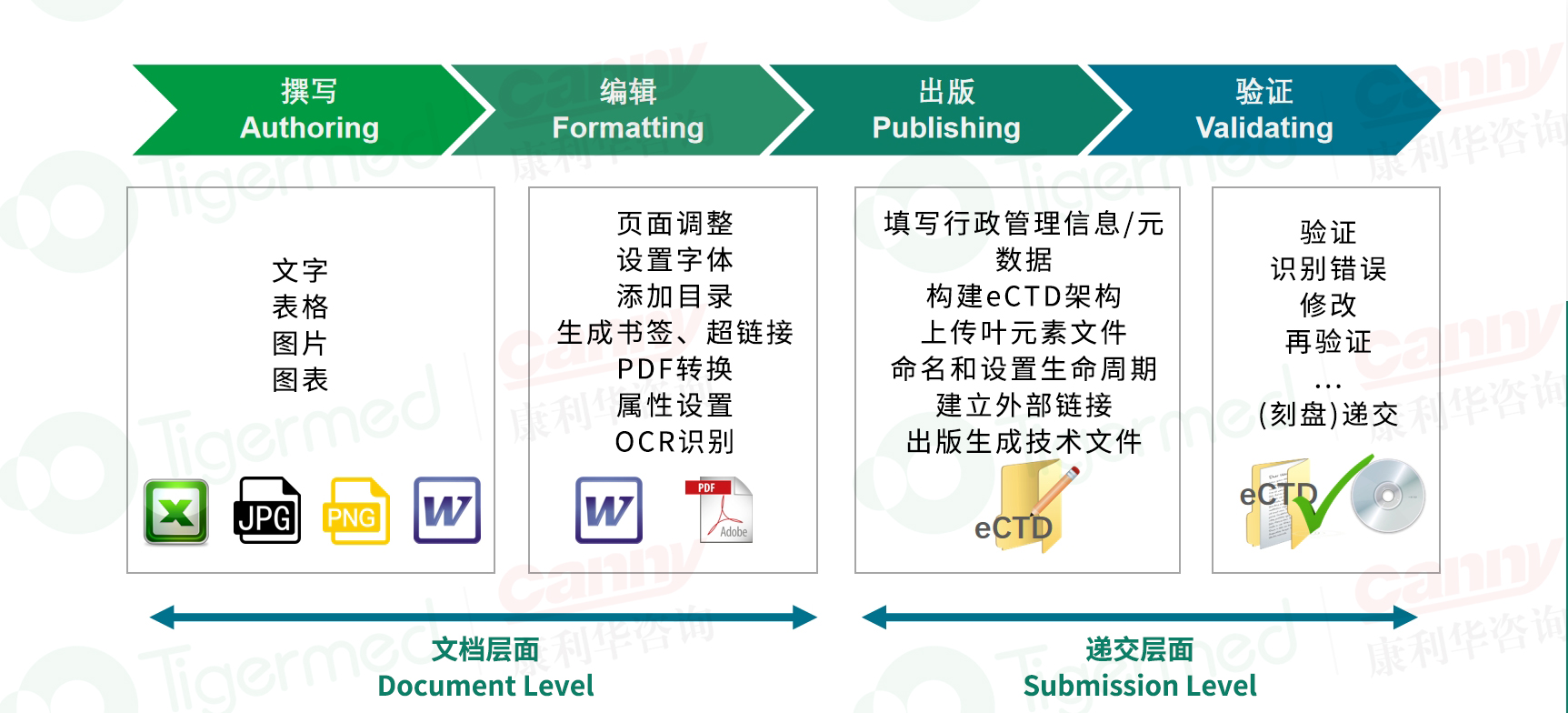
1)撰写:结构化文档开发:基于ICH CTD/eCTD规范,撰写符合模块化要求的技术文档(模块2-5),确保科学内容完整性与逻辑性。 监管合规性整合:嵌入元数据(如文档类型、序列号)、超链接及书签,满足目标机构(FDA/EMA/PMDA)的格式规范。;
2)编辑:技术审查:对内容进行科学准确性、一致性和合规性审核,包括数据交叉验证(如模块2摘要与模块3-5数据对齐)。 格式标准化:统一文档样式(字体、页眉/页脚、目录层级),优化PDF属性(兼容PDF/A标准),修复损坏文件或失效链接。
3)出版:XML骨干文件生成:使用专业工具构建XML导航结构,关联PDF文档与模块路径。 电子化封装:按eCTD目录结构(如M1-M5)组织文件,生成完整的eCTD提交包(含PDF、XML、PDF校验文件)。
4)验证:自动化合规检查:通过官方验证工具检测文件有效性及合规性。 人工复核:确认生命周期管理(序列号递增、删除操作标记)、地区差异化要求。
您值得信赖的医药法规符合专业顾问
全国热线:
400-8770626
邮箱:
Canny@TigermedGrp.com
地址:
中国·北京市朝阳区朝阳门外大街20号联合大厦808室
邮编:
100022

